Если очистка воды от грубодисперсных примесей (ГДП) может быть принципиально осуществлена обычным отстаиванием, время которого определяется размером и удельной массой частиц, то коллоидные примеси за счет их агрегативной устойчивости могут быть выделены из воды только методом коагуляции. Коагуляция - физико-химический процесс укрупнения коллоидных частиц за счет их слипания под действием молекулярных сил притяжения. В практике водоподготовки под коагуляцией понимают очистку воды от коллоидных веществ с одновременной очисткой от ГДП и обесцвечиванием воды путем дозировки в обрабатываемую воду специального реагента - коагулянта.
Для коллоидных систем характерны два основных признака: гетерогенность и дисперсность. Первый из признаков указывает на наличие межфазной поверхности (поверхностного слоя), определяющего самые существенные и характерные свойства коллоидных систем. Второй признак - дисперсность (раздробленность) - либо определяется по трем измерениям тела, либо характеризуется величиной, обратной минимальному размеру и названной дисперсностью, либо, наконец, используется третья характеристика раздробленности - удельная площадь поверхности Sуд (отношение межфазной поверхности к объему тела, табл. 3.1).
| Размер ребра кубика, см | Число частиц | Удельная площадь поверхности Sуд, см2/см3 |
Дисперсность, см-1 |
|---|---|---|---|
| 1 | 1 | 6 | 1 |
| 10-1/(мм) | 103 | 60 | 10 |
| 10-4(мкм) | 1012 | 6*104(6м2см3) | 104 |
| 10-7(нм) | 1021 | 6*107(6000м2/см3) | 107 |
Учитывая эти признаки, коллоидные системы можно охарактеризовать и определенным видом энергии, которым они обладают. Гетерогенность предопределяет наличие поверхностного натяжения Sг, причем чем сильнее различаются фазы по своей природе, тем выше поверхностное натяжение.
Коллоидные системы, обладающие большим избытком поверхностной энергии, стремятся уменьшить ее за счет снижения поверхностного натяжения благодаря поверхностной адсорбции других веществ, что формирует структуру поверхностного слоя и наделяет этот слой свойствами, отличными от свойств основной фазы. Избирательная адсорбция приводит к приобретению соприкасающимися фазами зарядов противоположного знака, но различной величины,т. е. возникает двойной электрический слой, обусловливающий различные электрокинетические явления.
Возможны три механизма образования двойного электрического слоя (ДЭС): во-первых, за счет перехода ионов или электронов из одной фазы в другую (поверхностная ионизация), во-вторых, благодаря адсорбции соединений - примесей водных сред и, в-третьих, за счет определенного ориентирования молекул сопряженных фаз в результате их взаимодействия. Для водных коллоидных систем характерны все три возможных вида ДЭС, причем роль каждого из них в значительной мере связана со значением рН среды и химическими свойствами коллоидных соединений. Так как все явления на межфазных поверхностях коллоидных систем обусловлены наличием ДЭС и количественные связи между коллоидными частицами зависят от его строения, то представляется важным оценить строение ДЭС.
Двойной электрический слой впервые рассмотрен Гельмгольцем в виде плоского конденсатора с расстоянием между двумя рядами разноименно заряженных ионов, близким к размеру диаметра молекул. На этом участке потенциал линейно снижался от значения ------(ро-------- до нуля. Однако такое строение слоя не учитывает теплового движения ионов. Гуи и Чепмен предположили, что ДЭС имеет размытое (диффузионное) строение, а концентрация противоионов (ионов, находящихся в растворе, со знаком заряда, противоположным заряду адсорбированных ионов и называемых потенциалообразующими) в поле действия поверхностного потенциала распределяется исходя из теплового движения в соответствии с законом Больцмана.
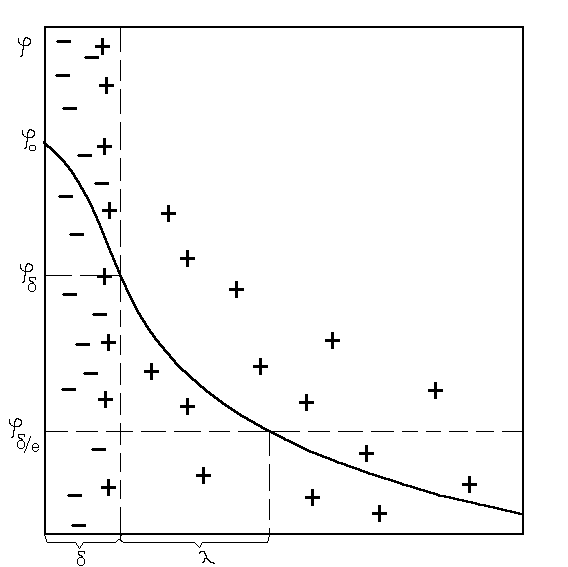
Современная теория строения ДЭС, логически развивающая представления Гельмгольца, Штерна, Гуи и Чепмена, рассматривает слой в виде двух частей (рис. 3.1). Одна его часть находится непосредственно у межфазной поверхности и образует адсорбционный слой (слой Гельмгольца) толщиной б, соответствующий диаметру гидратированных ионов, находящихся в нем.
Рис 3.1. Двойной электрический слой и изменение в нем потенциала
Остальная часть противоионов находится в диффузной части толщиной К, зависящей от свойства и состава раствора и коллоидной системы. Значение потенциала в слое Гельмгольца при удалении от потенциалообразующих ионов снижается линейно от фо до потенциала диффузного слоя ------(ре--------, а затем изменяется по экспоненте. Толщина диффузного слоя К соответствует, как было условно принято, расстоянию, на котором потенциал диффузного слоя снижается в е раз, т. е. в 2,718 раза.
Концентрация ионов в диффузном слое на расстоянии х от границ раздела фаз пропорциональна распределению Больцмана:
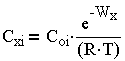
| 3.1 |
где С-концентрация иона; i, z- валентности ионов в объеме раствора за пределами диффузного слоя;
WX=zFфX характеризует работу по перемещению 1 моля ионов из объема раствора на расстояние X до
границы раздела фаз; F - число Фарадея; (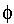 х - потенциал на расстоянии х,
изменяющийся от х до
х - потенциал на расстоянии х,
изменяющийся от х до  при х= и х=0 при до=
при х= и х=0 при до=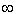 .
.
Использовав уравнение (3.1), можно оценить объемную плотность заряда р для сферического электрического поля в диффузном слое:
в присутствии одного электролита:
| Px=F(Z+CX++Z-CX-) | 3.2 |
в присутствии же нескольких электролитов:
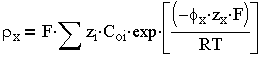
| 3.3 |
Для плоского двойного электрического слоя, радиус кривизны поверхности которого
значительно больше толщины ДЭС, соотношение между объемной плотностью заряда 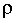 и потенциалом
определяется уравнением Пуассона по одной координате:
и потенциалом
определяется уравнением Пуассона по одной координате:
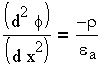
| 3.4 |
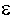 а - абсолютная диэлектрическая проницаемость. Подставляя (3.3) в (3.4), получаем уравнение
Пуассона - Больцмана, выражающее закон изменения поверхностного потенциала от расстояния в
диффузной части ДЭС и от свойств раствора
а - абсолютная диэлектрическая проницаемость. Подставляя (3.3) в (3.4), получаем уравнение
Пуассона - Больцмана, выражающее закон изменения поверхностного потенциала от расстояния в
диффузной части ДЭС и от свойств раствора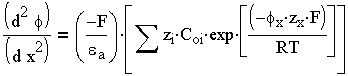
| 3.5 |
С помощью преобразований и решения этого уравнения можно численно оценить толщину диффузного
слоя  на границе раздела фаз. Так, значения
для симметричного одновалентного водного раствора электролита при значениях
=81, Т ==293 К, R=8,3Дж/(моль.К). F=9,64*104кл/моль и при
концентрациях С=10-1, 10-3 и 10-5моль/кг составляют
соответственно 1, 10, 100 нм.
на границе раздела фаз. Так, значения
для симметричного одновалентного водного раствора электролита при значениях
=81, Т ==293 К, R=8,3Дж/(моль.К). F=9,64*104кл/моль и при
концентрациях С=10-1, 10-3 и 10-5моль/кг составляют
соответственно 1, 10, 100 нм.
Кроме свойств раствора на формирование ДЭС у поверхности твердой фазы оказывает влияние ее природа.
В настоящее время сформировалось представление о том, что на поверхности оксидов, находящихся в водных
растворах, потенциалообразующими ионами ДЭС являются ионы Н+ или ОН- в зависимости
от рН среды и кислотно - основных свойств оксидов. Например, гидроксид кремния обладает ярко
выраженными кислотными свойствами, и поэтому при рН выше изоэлектрической точки [рН изоэлектрической
точки (рНиэ) - это то значение рН, при котором данное вещество не диссоциирует:рНиэSiO2
=2] его поверхность заряжена отрицательно:
[SiO2]m*nOH-(n- )H+
H+ )H+
H+
| 3.6 |
показывает долю диссоциированных молекул
вещества. Этот же результат можно получить, рассмотрев диссоциацию поверхностных молекул коллоидных
поликремниевых кислот:| (H2O)n(SiO2)m=H+
[(n-)H+nOH(SiO2)m]-
|
Подобным же образом возникает ДЭС и на поверхности между природной водой и органическими кислотами (гуминовыми, таниновыми и др.), для которых рН=(3.5-4.5), поэтому диссоциация их молекул поверхностного слоя при рН водной среды (6.5-8.0) протекает по кислотному характеру с возникновением отрицательного потенциала.
Гидроксид железа имеет основные свойства, что определяет положительный заряд его поверхности, так же как и органических оснований (аминов).
Для амфотерных гидроксидов, например для гидроксида алюминия, значительно влияние рН среды на изменение и знака заряда поверхности: так, в кислой и нейтральной средах гидроксид алюминия (рНИЭ =7.6 - 8.2) имеет положительно заряженную поверхность, а в щелочной среде (при рН>рНИЭ) она заряжена отрицательно.
В коллоидных системах ДЭС возникает на поверхности частиц. Частицу дисперсной фазы вместе с ДЭС называют
мицеллой. Внутреннюю часть мицеллы составляет агрегат основного вещества {[SiO2]m},
на поверхности которого расположены потенциалообразующие ионы {ОН-}. Агрегат с
потенциалообразующими ионами образуют ядро мицеллы {[SiO2]mnOH-}.
Ядро с противоионами плотной части ДЭС составляет гранулу {[Si02]mnOH-
(n-)H+}. Заряженная гранула, окруженная противоионами диффузного
слоя, представляется электронейтральной мицеллой {[SiO2]mnOH-
(n-)H+H+}.
При тепловом движении молекул коллоидные частицы вовлекаются в молекулярно-кинетическое движение среды. Это явление было обнаружено в 1927 г. английским ботаником Робертом Броуном, наблюдавшим в микроскоп непрерывное движение (броуновское движение) очень мелких частичек - спор цветочной пыльцы, взвешенных в воде. В настоящее время теоретически и экспериментально доказано, что молекулы среды (жидкости или газа) сталкиваются с частицей дисперсной фазы, в результате чего она получает огромное число ударов со всех сторон (более 1020 толчков/с). При больших размерах частицы результирующий импульс огромного числа ударов со всех сторон согласно соответствующему закону статически оказывается равным нулю, и частица большой массы и инерционности не будет двигаться под действием теплового движения молекул. Очень малые частицы имеют значительно меньшую массу и поверхность, на которую будет приходиться гораздо меньшее число ударов, поэтому увеличивается вероятность неравномерности распределения импульсов с разных сторон, в результате чего частица приобретает колебательное и поступательное движение.
Эйнштейн (1905 г.) и Смолуховский (1906г.) независимо друг от друга, постулируя единство броуновского и молекулярно-кинетического движений, установили количественную связь между средним сдвигом частицы А (называемым иногда амплитудой смещения) и коэффициентом диффузии D за время r:
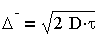
| 3.7 |
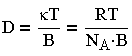
| 3.8 |
где k-постоянная Больцмана; Na - число Авогадро; В - коэффициент трения; Т - температура.
Уравнение (3.7) в предположении применения закона Стокса к движению частиц:
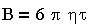
| 3.9 |
трансформируется к виду:
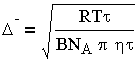
| 3.10 |
Из уравнения (3.10) следует, что частицы перемещаются тем быстрее, чем меньше размер частиц г, вязкость жидкой фазы и чем выше температура.
При движении коллоидных частиц в жидкости, т. е. при относительном перемещении фаз, происходит разрыв ДЭС по плоскости скольжения. Часть диффузного слоя захватывает частица, а часть ионов остаются в растворе, в результате чего устанавливается разность потенциалов между движущейся коллоидной частицей и окружающей ее средой на поверхности скольжения при отрыве части диффузного слоя. Потенциал на поверхности скольжения называется электрокинетическим, потенциалом или s (дзета) - потенциалом.
Значение дпотенциала всегда меньше потенциала ДЭС, и это различие тем больше, чем меньше толщина диффузной части слоя (рис. 3.2). Поэтому такие факторы, как понижение температуры, введение в раствор электролита (C3>C2>C1), не взаимодействующего с поверхностью, а также увеличение заряда его ионов, снижая толщину диффузного слоя, уменьшают электрокинетический потенциал.
Электрокинетические характеристики системы зависят в большой степени от свойств поверхности контактирующих фаз и рН среды, поскольку ионы H+ и ОН- обладают высокой специфической адсорбционной способностью.
Так, многие неорганические оксиды (кремния, железа, алюминия) характеризуются активной поверхностью, содержащей ионогенные группы, диссоциация которых в зависимости от рН раствора, как отмечалось, определяет заряд поверхности. Потенциал таких коллоидных оксидов может достигать 100 мВ, а при изменении рН среды в интервале выше и ниже значения рНиэ возможна перезарядка фазы.
На инертных поверхностях (графите, масле и др.), лишенных ионогенных групп, заряд возникает в результате специфической адсорбции ионов.
Экспериментально значение электрокинетического потенциала определяют методом электрофореза, используя
модифицированное расчетное уравнение Гельмгольца - Смолуховского:
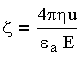
| 3.11 |
где u - средняя линейная скорость движения частиц; Е - градиент потенциала внешнего электрического поля.
Отношение u/Е при электрофорезе называют электрофоретической подвижностью и эф. Для расчета потенциала,
В, на коллоидных частицах, находящихся в разбавленных водных растворах при 20°С, используют соотношение:
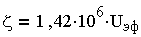
| 3.12 |
где UЭф выражена в м2/(c*В); опытные значения UЭф достигают 5*10-8
м2/(c*В), а в соответствии с (3.12) -потенциал коллоидов
природных вод составляет около 70 мВ.
Коллоидные системы в природных водах имеют обычно одинаковый отрицательный заряд и характеризуются агрегативной устойчивостью, т.е. стабильностью дисперсности, распределением по объему и взаимодействием между частицами. Потеря агрегативной устойчивости дисперсной системой, заключающаяся в слипании частиц, представляет собой, как отмечалось, коагуляцию. которая приводит в конечном счете к расслоению фаз.
Схема переходов дисперсных систем в различные состояния представлена на рис. 3.3. Устойчивая дисперсная фаза, равномерно распределенная по всему объему, может образовываться в результате конденсации из истинного раствора. Потеря агрегативной устойчивости при воздействии на систему - первый этап коагуляции заключается в сближении коллоидных частиц и взаимной их фиксации на небольших расстояниях, разделенных прослойками среды, с образованием так называемых флокул, в которых частицы обладают относительной подвижностью при воздействии относительно небольших сил. Обратный процесс разрушения флокул называется пептизацией.
Рис 3.3. Схема процессов протекающих в дисперсных системах
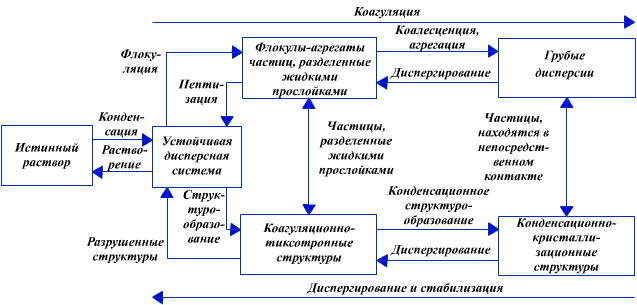
Более глубокий процесс коагуляции приводит к разрушению прослоек жидкости и непосредственному контакту частиц с образованием жестких структур из твердых частиц (или слиянию жидких, например нефтепродуктов, называемому коалесценцией). Разрушения образовавшихся структур можно достичь только принудительным диспергированием. Таким образом, в ходе коагуляции осуществляется ряд процессов структурообразования, протекающих с уменьшением удельной поверхности системы. Каждая промежуточная структура имеет свои свойства.
При рассмотрении агрегативной устойчивости и скорости коагуляции коллоидных систем различают ряд факторов устойчивости. В реальных системах действуют смешанные факторы, проявляющиеся как совокупность термодинамических (снижающих межфазное натяжение) и кинетических факторов (проявление свойств межчастичных прослоек).
Несмотря на отсутствие количественной теории агрегативной устойчивости, в настоящее время разработана достаточно детально электростатическая теория устойчивости, ряд положений которой рассматривается ниже.
Между коллоидными частицами одного знака заряда (содержащиеся в природной воде органические примеси, в том числе железосодержащие, и коллоидная кремнекислота заряжены отрицательно) действуют молекулярные силы притяжения (силы Ван-дер-Ваальса) и электростатические силы отталкивания (кулоновское взаимодействие). Таким образом, стабильность (устойчивость) дисперсной фазы и ее коагуляция определяются соотношением между силами притяжения и отталкивания частиц.
В наиболее общем виде теория взаимодействия частиц была разработана советскими учеными Б.В.Дерягиным и Л.Д.Ландау в 1937-1941 гг. и несколько позже независимо от них голландскими учеными Фервеем и Овербеком. По первым буквам фамилий этих ученых теория взаимодействия и коагуляции дисперсных частиц получила сокращенное название ДЛФО. Простейший случай этой теории рассматривает взаимодействие без учета теплового движения двух крупных частиц, у которых толщина ДЭС значительно меньше размера частиц, что позволяет перейти к оценке взаимодействия двух плоских параллельных пластин.
Суммарная энергия взаимодействия пластин, приходящаяся на единицу площади, складывается из энергии электростатического отталкивания и молекулярного притяжения UМ:
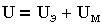
| 3.13 |
Количество энергий зависит от расстояния h между пластинами:
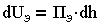
| 3.14 |
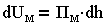
| 3.15 |
где Пэ, Пм - давления отталкивания и притяжения, являющиеся составляющими расклинивающего давления.
Понятие расклинивающего давления в теории агрегативной устойчивости было введено Б. В. Дерягиным в 1935 г. Оно возникает при сильном уменьшении толщины пленки при взаимодействии сближающихся поверхностных слоев и обусловливается сближением как фаз так и межфазных слоев. Расклинивающее давление можно рассматривать как избыточное давление, которое нужно приложить к пленке, чтобы сохранить ее равновесную толщину, поэтому отталкивание поверхностных слоев (Пэ) рассматривается как положительное, а притяжение (сжатие пленки Пм) - как отрицательное.
В теории ДЛФО принято, что давление отталкивания характеризуется только электростатическими силами, поэтому
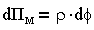
| 3.16 |
В соответствии с теорией Гуи - Чепмена изменение потенциала (рис. 3.1) соответствует уравнению:
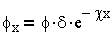
| 3.17 |
Интегрирование уравнения (3.16) в пределах от 2
X до 0 и дальнейшее интегрирование уравнения (3.14) в пределах от h (расстояние между пластинами)
до бесконечности позволяют выразить в окончательном виде:
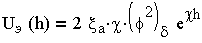
| 3.18 |
Из (3.18) следует, что UЭ(h) уменьшается с увеличением расстояния по экспоненциальному закону.
Энергия притяжения частиц в теории ДЛФО определялась суммированием взаимодействий между атомами и молекулами. В основу вывода этого уравнения положено уравнение адсорбционного притяжения (Uадс) одной молекулы к поверхности адсорбента (в данном случае частицы). Энергия молекулярного притяжения пластин в среде описывается уравнением: table>
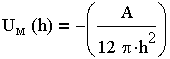
| 3.19 |
где A - константа Гамакера. Эта константа учитывает природу взаимодействующих тел, выражается в единицах энергии (порядка 10-19 Дж) и слагается из отдельных констант, характеризующих свойства дисперсной фазы и среды.
Уравнение (3.19) показывает, что энергия притяжения пластин (частиц) обратна квадрату расстояния между ними, т. е. она уменьшается значительно медленнее с расстоянием, чем энергия притяжения между молекулами (атомами), которая обратно пропорциональна расстоянию в шестой степени. Отсюда следует, что коллоидные частицы взаимодействуют на более далеких расстояниях, чем молекулы.
Зная закономерности для энергии отталкивания и энергии притяжения частиц, простым сложением получаем общую удельную энергию взаимодействия между параллельными пластинами (частицами):
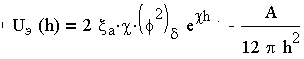
| 3.20 |
Энергия взаимодействия сферических частиц вычисляется по более сложным уравнениям, примером которых может служить уравнение (3.21). полученное для частиц с низким потенциалом (характерным для коллоидных систем природных вод) и радиусом частиц существенно превышающим толщину диффузного слоя :
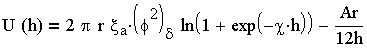
| 3.21 |
Уравнение (3.20), полученное в соответствии с теорией ДЛФО, характеризует поведение дисперсных систем, устойчивость которых или коагуляция определяется значением и знаком общей потенциальной энергии взаимодействия частиц. Характер изменения энергии отталкивания Uэ(h) и энергии притяжения Uм(h) показан на рис.3.4.
Рис 3.4. Потенциальные кривые взаимодействия коллоидных частиц
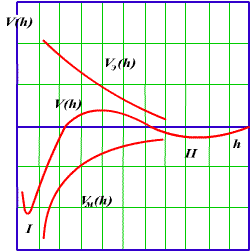
Результирующая кривая u(h) получена путем геометрического сложения соответствующих
ординат. Напомним, что положительная энергия отталкивания уменьшается с увеличением расстояния по
экспоненциальному закону, а отрицательная энергия притяжения Uм(h) обратно пропорциональна квадрату
расстояния. В результате при h>0 иэ Uэ-const, Uм=-оо и при больших расстояниях, когда экспонента
убывает значительно быстрее, чем степенная функция, между частицами преобладает энергия притяжения
(I и II энергетические минимумы), а на средних расстояниях, соответствующих толщине ДЭС,-энергия
отталкивания, представляющая собой потенциальный барьер, препятствующий слипанию частиц.
Потенциальный барьер возрастает с увеличением [см. (3.21)], и, как показывает практика,
при =20мВ образуется потенциальный барьер, обеспечивающий устойчивость
дисперсной системы. Коллоидные системы природных вод в соответствии с (3.12) имеют =70мВ.
С учетом того, что > (т.е. больше 70 мВ),
они характеризуются высоким потенциальным барьером и большой агрегативной устойчивостью.
В рассмотренном упрощенном варианте теории ДЛФО не учитывается размер частиц и их форма, однако с учетом уравнения взаимодействия сферических частиц (3.21) можно видеть, что высота электростатического барьера и, как следствие, устойчивость к коагуляции повышаются с ростом радиуса частиц (в первом приближении пропорционально радиусу). Одновременно с этим увеличение размера частиц приводит к увеличению второго энергетического минимума, что подтверждается наличием процессов дальней агрегации в грубодисперсных системах.
Хотя электростатическая теория устойчивости дисперсных систем рассматривает лишь один электростатический фактор устойчивости, этот фактор наиболее характерен для коллоидных систем в водных средах, создающих условия для их стабильности.
Теория ДЛФО позволяет утверждать, что все факторы, которые определяют высоту энергетического барьера, зависящую от электрического потенциала, и толщину ДЭС (поверхностная диссоциация, количество адсорбированных потенциалообразующих ионов и прочность их закрепления, а также взаимодействия противоионов с поверхностью, определяющие толщину ДЭС), играют основную роль в агрегативной устойчивости реальных коллоидных систем. Уменьшение же потенциала или сжатие диффузной части ДЭС приведет к протеканию соответственно нейтрализационной или концентрационной коагуляции.
Это подтверждено многочисленными экспериментальными и промышленными данными. Однако в практике водоподготовки осуществление коагуляционных процессов с использованием электролитов для уменьшения дзета - потенциала приводит к необходимости снижения рН при коагуляции природных вод и повышению их солесодержания, что недопустимо по технико - экономическим соображениям с учетом последующей стадии полного или частичного удаления ионизированных примесей из обрабатываемой воды. При подготовке добавочной воды используют вариант коагуляции, связанный с взаимной коагуляцией разнородных дисперсных систем - гетерокоагуляцией. Гетерокоагуляция происходит тем эффективнее, чем полнее произойдет нейтрализация зарядов частиц в смешиваемых дисперсных системах с противоположными знаками заряда коллоидных частиц природных вод (обычно заряженных отрицательно) и искусственно созданной коллоидной системой с положительным зарядом. Предварительная очистка природных и сточных вод от коллоидных примесей методом коагуляции производится добавлением минеральных солей с гидроизолирующими катионами (коагулянтов), анодным растворением металлов или изменением рН среды в тех случаях, когда в сточных водах содержатся в достаточном количестве катионы, способные при гидролизе образовывать труднорастворимые соединения. В качестве коагулянтов применяются обычно сульфат алюминия [А12(S04)3* 18Н20] или сульфат железа (FeS04*7H20), причем последний реагент используют при совмещении процессов коагуляции и известкования (содоизвесткования). Это связано с тем, что окисление Fe2+ кислородом, растворенным в обрабатываемой воде, происходит медленно и скорость окисления до-стигает практически приемлемого значения лишь при рН>8 (рис. 3.5).
Рис 3.5.Зависимость времени окисления двухвалентного железа кислородом от pH воды
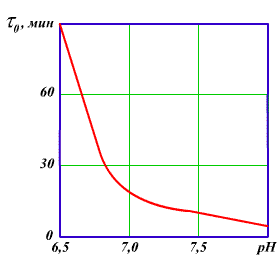
Необходимость окисления и, следовательно, достижения полноты гидролиза вытекает из сопоставления произведений растворимости гидроксидов металлов, значения которых при температуре 18°С для Fе(ОН)3 равны 3.8-10-38, Fe(OH)2 4.8-10-16 и для А1(ОН)з 1,9-10-33.
| Al2(SO4)3=2Al3++3SO2-4 | 3.22 |
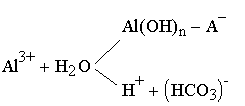
| 3.23 |
Схема включает наряду с диссоциацией и собственно гидролизом три сопутствующие реакции: 1) нейтрализацию ионов водорода, образовавшихся при гидролизе (Н++НСО3-); 2) формирование гидроксидбикарбонатных соединений алюминия [А1(ОН)n + НСО3-] из-за конкуренции ионоНСО3- и ОН- за координационные участки А13+; 3) возникновение основных солей алюминия с другими анионами воды [А1(ОН)- + А)-], например оксисульфатов типа A10HS0)-, за счет поверхностных ионообменных реакций. Состояние рассматриваемой системы характеризуется определенным динамическим равновесием, связанным с условиями обработки воды.
При щелочности воды более 1.0-1.5 мг-экв/кг не возникает затруднения в отводе из сферы реакции образующихся катионов Н+, поэтому разность между щелочностями исходной и обработанной воды близка к расчетной. В этом случае щелочность уменьшается на значение, равное дозе коагулянта. В тех случаях, когда щелочность обрабатываемой воды недостаточна для связывания образующихся при гидролизе коагулянта ионов Н+ (например, в паводковый период), необходимо воду подщелачивать для достижения полноты гидролиза всего введенного коагулянта.
После ввода коагулянта в обрабатываемой воде через несколько минут появляются хлопья белого или желтого цвета, однако до образования видимых хлопьев частицы А1(ОН)3 проходят коллоидную фазу дисперсности и характеризуются положительным знаком заряда. На этой скрытой стадии коагуляции происходит главным образом сложный комплекс процессов очистки воды от коллоидных примесей: взаимная коагуляция разноименно заряженных коллоидов при взаимодействии дестабилизированными участками поверхности (гетерокоагуляции), электролитная коагуляция под влиянием ионов SO42- и образующихся в первый момент ионов А3+, способствующих сжатию ДЭС коллоидных частиц. В дальнейшем микрохлопья сцепляются, захватывая грубодисперсные примеси и воду и образуя коагуляционную структуру в виде хлопьев (флокул) размером 0,5-3 мм с плотностью 1,001-1,1 т/м3 (рис. 3.6).
Рис 3.6.Схема кофгуляционной структуры
Количественная теория кинетики коагуляции была создана М.Смолуховским, рассмотревшим скорость коагуляции монодисперсных золей со сферическими частицами, которые сталкиваются между собой под действием броуновского движения. Критическое расстояние взаимодействия частиц принято равным сумме радиусов частиц, что соответствует их непосредственному контакту. Согласно представлению М. Смолуховского взаимодействия происходят только между двумя частицами - одиночными, одиночными с двойными, двойными друг с другом и т. д., что позволяет представить процесс коагуляции аналогичным протеканию бимолекулярных химических реакций и использовать соответствующий математический аппарат.
Для скорости увеличения числа fix скоагулированных частиц, зависящей от начальной концентрации частиц По, интенсивности их броуновского движения, радиуса действия сил притяжения и эффективности соударений, Смолуховским получено следующее выражение, соответствующее реакции второго порядка:
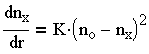
| 3.24 |
в котором К - константа скорости коагуляции, выраженная в общем виде пропорциональной распределению Больцмана:
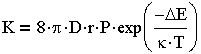
| 3.25 |
где D - коэффициент диффузии одиночных частиц; г-радиус частицы; Р-стерический фактор, учитывающий
расположение частиц при столкновении, их форму, размеры; k - постоянная Больцмана;  E - средняя энергия,
необходимая для слипания частиц (потенциальный барьер).
E - средняя энергия,
необходимая для слипания частиц (потенциальный барьер).
В теории коагуляции различают быструю и медленную коагуляцию. При быстрой коагуляции все столкновения
частиц эффективны, т.е. приводят к слипанию частиц, что отвечает условию E=О
и равенства единице стерического множителя р=1. Константа скорости быстрой коагуляции в соответствии с
(3.25) равна:
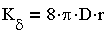
| 3.26 |
Для медленной коагуляции, при которой не все столкновения эффективны, E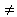 0 и Р0
Исходя из (3.25) и (3.26), константу скорости медленной коагуляции можно выразить так:
0 и Р0
Исходя из (3.25) и (3.26), константу скорости медленной коагуляции можно выразить так:
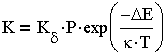
| 3.27 |
Отношение К/КM иногда называют фактором устойчивости или
стабильности системы. Если E значительно больше kT, то скорость коагуляции
может приблизиться к нулю и коллоидная система будет агрегативно - устойчивой.
Возвратившись к (3.24) и обозначив nо - nх в единице объема за временный интервал r через nr, получим:
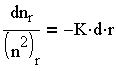
| 3.28 |
и после интегрирования выражения (3.28) имеем:
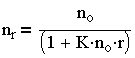
| 3.29 |
Из - за трудности теоретического определения константы скорости коагуляции Смолуховский ввел понятие периода половинной коагуляции (времени уменьшения концентрации частиц до половины от начальной). Из (3.29) следует:
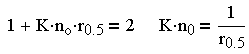
| 3.30 |
и
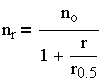
| 3.31 |
Выражение (3.31), несмотря на известные допущения в теории М. Смолуховского, нашло экспериментальное подтверждение и широко используется для обработки экспериментальных данных по кинетике коагуляции в монодисперсных системах.
В реальных полидисперсных системах коагуляция протекает быстрее: крупные частицы способствуют коагуляции более мелких. Экспериментально было установлено, что скорость объединения относительно мелких частиц (7-11 мкм) с более крупными (60 мкм) в турбулентном потоке была на несколько порядков выше, чем скорость коагуляции только мелких частиц. Этот факт объясняется различием в механизме встречи частиц - инерционным в случае полидисперсной системы и диффузионным в случае монодисперсной.
Практика эксплуатации водоочистных установок свидетельствует о том, что при любых исходных условиях полнота удаления коллоидных и грубодисперсных частиц в осветлителях при коагуляции зависит от оптимальной скорости, создаваемой при перемешивании воды в камере хлопьеобразования. Причем для каждой заданной скорости устанавливается равновесие между вероятностью образования хлопьев и их средним размером.
примечание